Algorithm Explanation
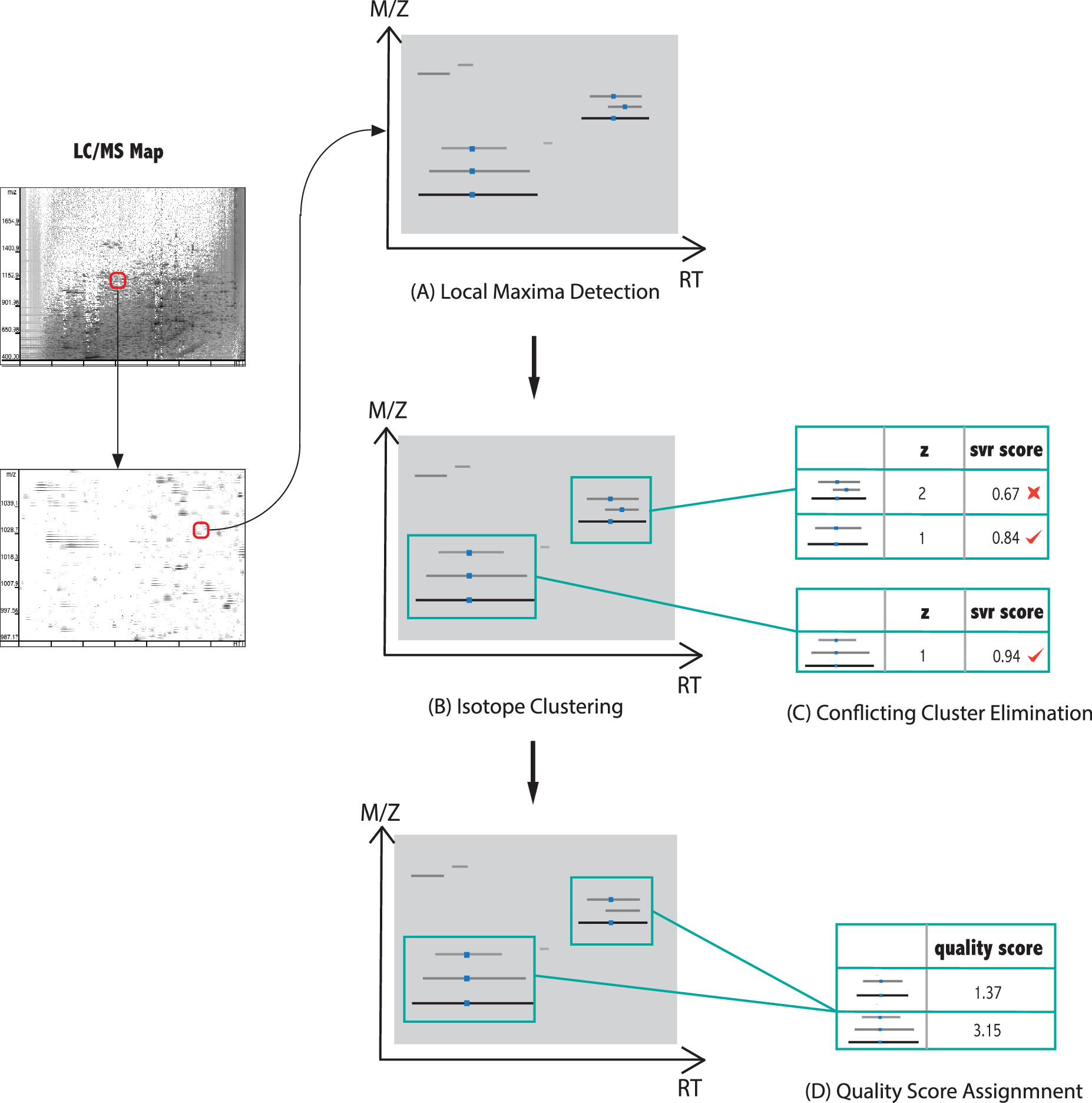
Step A: Detection of local maxima on trails
A trail consists of the signal peaks of the same isotopic precursor ion that appear persistently at the same m/z (allowing for a mass error tolerance) for a period of time. The signal intensity over time on a trail can be represented as the function h(t). By locating the most intensive peak signal on each trail, a precursor ion (either a monoisotope or an isotope) can be distinguished with confidence.
Step B: Grouping trails into isotope groups by m/z difference
We define an isotope group as a group of isotopes formed by clustering all the isotopes (represented by local maxima) for a monoisotope. A sequence of local maxima forms an isotope group if they have similar RT and the m/z of every pair of adjacent local maxima differ by m(proton)/k for an integer k. Here, k is the charge state of the candidate, and m(proton) is the atomic mass of a proton. We start searching for isotope groups from k = 1, 2, ..., N, where N is the largest charge state.
Step C: Eliminating conflicting isotope groups by SVR score
Isotope groups are candidates for peptide features. However, different isotope groups may conflict with each other. We have to determine the isotope group that the trail is most likely to belong to and remove the group in which the trail is falsely clustered. For example, the local maxima in each isotope group with charge 2k contain a subset of local maxima that form a charge-k isotope group. Allowing both charge states to exist will substantially increase the false positives. Therefore, a scoring function is used to remove the inferior isotope groups within a set of conflicting ones. After this filtration, the remaining isotope groups become the peptide features.
Step D: The assignment of the final NN-based quality score.
NN-based scoring function is used to assess the quality of each peptide feature. Finally, features with very close m/z and RT values and the same k are clustered to remove redundancies.